
The FDA has
approved KRAZATI

We’re tackling cancer head-on
At Mirati, we convert possibility into reality by developing best-in-class targeted therapies to improve the lives of patients.

At Mirati, we convert possibility into reality by developing best-in-class targeted therapies to improve the lives of patients.
Our Programs
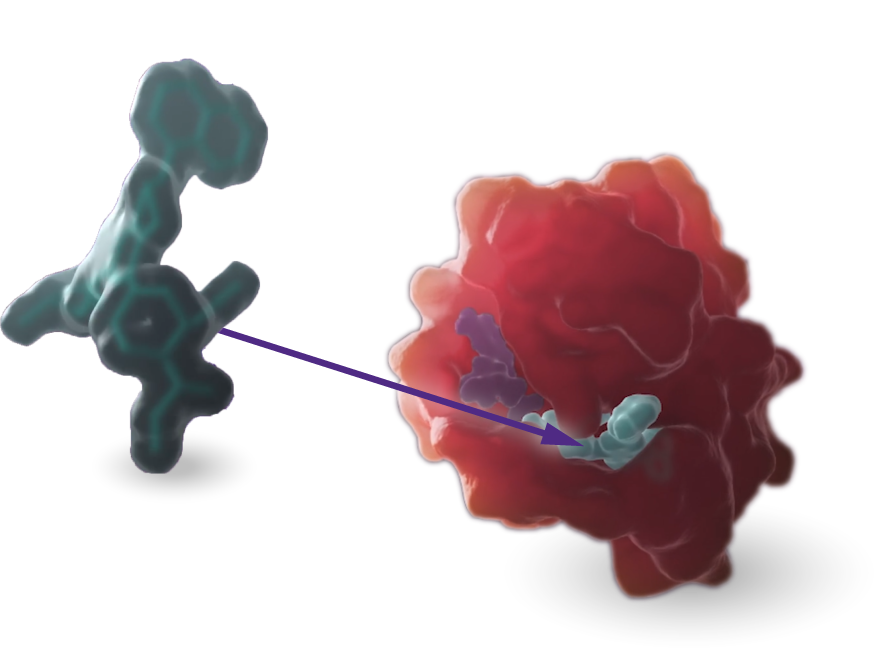
KRAS
Our scientists are rigorously advancing research and development programs targeting the KRASG12C and KRASG12D mutations.
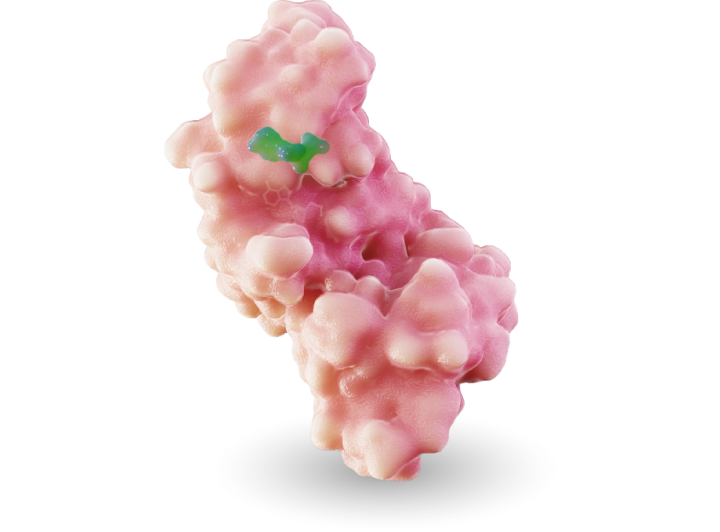
MRTX1719
MRTX1719 is an investigational, internally discovered synthetic lethal PRMT5 inhibitor for the treatment of methylthioadenosine phosphoylase (MTAP)-deleted cancers.
Mirati is developing a pipeline of novel therapeutics.

Become part of something bigger.
Error: Failed to decode JSON.